
Recreating gut-liver axis during NAFLD onset by using a Caco-2/HepG2 co-culture system

 , ...
, ... Abstract
Nonalcoholic fatty liver disease (NAFLD) onset and its progression towards nonalcoholic steatohepatitis (NASH) features increased intestinal permeability and leaky gut, thereby favoring the escape of endotoxin [lipopolysaccharides (LPS)] from the gut to the liver. The aim of this study was to resemble the crosstalk between intestine and liver during NAFLD by using an in vitro model of co-culture system. Enterocytes (Caco-2) were seeded on Transwell filters (pore size: 0.4 μm) and cultured for 21 days to constitute a confluent monolayer, and then they were co-cultivated with hepatocytes (HepG2) for an additional 24 h. Caco-2 on the apical chamber were exposed to LPS and/or a mixture of palmitic and oleic acid (PAOA) for 24 h. FITC-4000 dextrans (FD4) permeability across Caco-2 monolayer was increased by the treatment of Caco-2 cells with PAOA and LPS, consistently with tight junction-associated proteins reduction. Caco-2 exposure to PAOA/LPS promoted ApoB, triglyceride (TG), and free fatty acid secretion in basolateral media. In turn, HepG2 co-cultured with Caco-2 exposed to LPS, PAOA, or both accumulated lipid droplets and increased intracellular TG content. Likewise, Caco-2 released pro-inflammatory cytokines in basolateral media. These events triggered endoplasmic reticulum (ER) and oxidative stress, enhancing reactive oxygen species (ROS), H2O2, aldehyde derivate production, and ROS-induced DNA damage in HepG2 cells. Hence, Caco-2/HepG2 co-culture system may faithfully reproduce the breach in the intestinal barrier integrity that occurs in NAFLD, thus resulting in the increased inflammatory response and ER and oxidative and stress, which promote the switch towards NASH.
Keywords
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD), or as it has been more recently re-named “metabolic dysfunction-associated fatty liver disease (MAFLD)”[1], is the most frequent chronic hepatic disorder worldwide, affecting 20%-40% of the adult population in Western countries[2,3]. Hence, due to its ever rising prevalence, it constitutes a huge socioeconomic burden[4]. NAFLD is diagnosed when hepatic fat accumulation exceeds 5% of liver weight in absence of alcohol misuse, and it entails a wide phenotypic variety of hepatic aberrancies, ranging from simple and uncomplicated steatosis to nonalcoholic steatohepatitis (NASH), in which steatosis is intertwined with inflammation, oxidative stress, and hepatocyte degeneration. This condition may be aggravated by fibrosis, and, in some cases, it may worsen into cirrhosis and hepatocellular carcinoma (HCC)[5,6]. The convoluted natural course of the disease relies on multiple drivers, encompassing exacerbated immune system activation, endoplasmic reticulum (ER) and oxidative stress, organelle impairment, and DNA derangements[7]. However, the fine-tuned mechanisms which regulate the switching from fatty liver to NASH are still under study.
In this context, mounting evidence points to the gut-liver axis as one of the primary contributors to NAFLD development and its precipitation to NASH up to more severe forms, as a consequence of the close anatomical and functional relationship between the gastrointestinal tract and the liver[8,9]. Indeed, the hepatic tissue is exposed to a perpetual flow of potentially harmful gut microbial byproducts and ingested nutrients via the portal vein, and, consequently, the liver modulates microbiota taxonomic composition through bile acid secretion into the duodenum[8]. The bowel wall displays immunological and selective barrier properties, regulating bidirectional fluxes between the gut and the liver, due to the presence of adherent and tight junctions [constituted by zonula occludens 1 (ZO-1) and claudins] and desmosomes, which hold together the juxtaposed epithelial cells and protects the host from invasive bacteria, thus avoiding pathogens overgrowth and their systemic translocation[8,10]. Notwithstanding, defects in intestinal barrier integrity (a phenomenon known as leaky gut) along with imbalances in gut microbiota proportion (referred to as dysbiosis) frequently affect patients with severe NAFLD[11,12]. As a result, enhanced gut permeability has been associated with the escape of pathogenic microorganisms and endotoxemia, thereby fostering intestinal mucosal inflammation and a chronic low-grade immune system activation, as reported in several studies[13-16]. Therefore, disturbances in the gut-liver axis homeostasis may describe the evolution of steatosis to NASH and HCC[17-21]. Lipopolysaccharides (LPS) per se, including several pathogen-associated molecular patterns, activate toll-like receptor (TLR) cascade in hepatocytes, Kupffer cells, and hepatic stellate cells, elevating serum tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL6) and precipitating systemic inflammation and fibrosis[22,23]. Otherwise, exaggerated amounts of free fatty acids (FFAs), whose levels are typically increased in NAFLD due to enhanced adipose tissue lipolysis, may independently modulate inflammatory pathways[24,25]. In turn, severe inflammation in NASH patients may contribute to favor intestinal high-permeability and dysbiosis[26]. In addition, similar to pro-inflammatory mediators, microRNAs (miRNAs) and dysregulated bile acid secretion may also influence gut barrier integrity by targeting ZO-1, claudins, and occludins[27]. However, intensive efforts are essential to completely understand the true causality between intestinal impairments and hepatic morbidities as well as to pinpoint the mechanisms through which intestinal alterations may affect liver pathology.
Thus, to model the interaction which occurs between the gut and the liver in the context of NAFLD, a representative in vitro co-culture system is necessary. Unfortunately, these studies may often require sophisticated and expensive intestine-liver-on-chip platforms[28], partially limiting their wide applications. Here, our aim was to exploit a reproducible protocol to possibly integrate different signals that participate to the onset of NAFLD and its progression towards NASH. Since increased intestinal permeability along with systemic LPS translocation represents the hallmarks of progressive NAFLD, featuring excessive FFA efflux to the liver, we aimed to recreate the gut-liver axis interactions by exploiting an in vitro model of a co-culture system of enterocytes (Caco-2) and hepatocytes (HepG2). Caco-2 on the apical chamber were exposed to LPS and/or a mixture of palmitic and oleic acid (PAOA) to recapitulate what occurs in NAFLD due to the presence of dysbiosis and higher fat intake. Then, we measured intestinal permeability and lipid and cytokine secretion in basolateral media. As concern the hepatic response, it was evaluated in terms of fat overload, inflammation, and ER and oxidative stress.
MATERIALS AND METHODS
Reagents
Dulbecco’s modified Eagle’s medium (DMEM), phosphate-buffered saline (PBS), fetal bovine serum (FBS), L-glutamine, penicillin/streptomycin, Trypsin/EDTA, Hank’s balanced salt solution, and fast SYBR green master mix were obtained from ThermoFisher Scientific (Waltham, USA). Cell culture flasks, plates, and polyester membrane inserts (12 mm diameter, 0.4 μm pore diameter) were obtained from Corning (Cambridge, MA, USA). Free fatty acids (FFAs) Quantification Kit and Triglyceride Quantification Kit were purchased from BioVision (Milpitas, United States). Cholesterol Colorimetric Assay Kit - HDL and LDL/VLDL, DCF ROS/RNS Colorimetric Assay Kit, Hydrogen Peroxide Assay Kit, Lipid Peroxidation (MDA) Assay Kit, and DNA Damage Assay Kit were purchased from Abcam (Cambridge, UK). Anti-pTLR4, TLR4, ZO-1, and Claudin antibodies were acquired from Cell Signaling Technologies (Boston, USA). Anti β-Actin was purchased from Abcam (Cambridge, UK).
Cell culture and co-culture system
Caco-2 cells, a model of the human colon epithelial cancer cell line, were obtained from ATCC (HTB-37, LGC Standards, Milan, Italy) and were cultured at 50% confluency and maintained at 37 °C under 5% CO2 atmosphere in DMEM containing 1% L-glutamine, 100 U/L penicillin, and 100 μg/L streptomycin, supplemented with 10% FBS. For Caco-2 differentiation, cells were seeded on polycarbonate filter Transwells with 12 mm of diameter and 0.4 μm semipermeable pore diameter (Transwell, Corning Inc., Lowell, MA, USA) at a 3.5 × 105 cells/cm2 density in complete medium supplemented with 10% FBS in both apical and basolateral compartments for two days to allow the formation of a confluent cell monolayer. Then, from the third day after seeding, fresh complete medium was added in both compartments and cells were cultured for 21 days with regular medium changes three times per week. During these 21 days, Caco-2 spontaneously differentiated into small intestinal phenotype, acquiring microvilli at the apical pore and formed tight intercellular junctions at the basolateral surface, thus mimicking the human intestinal absorption[29].
The HepG2 cell line was provided by from ATCC (HB-8065, LGC Standards, Milan, Italy), and it was cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 1% L-glutamine and incubated at 37 °C under 5% CO2 atmosphere. Caco-2 and HepG2 cells were used for no more than 20 passages after thawing, as the increase in the number of passages may affect cell phenotype and alter experimental results.
Cell monolayer integrity and differentiation evaluation
To determine whether Caco-2 cell layers were fully confluent, Transwells were placed in new multi-well plates and then phenol red leakage, through the intercellular spaces, from the apical to the basolateral compartment was examined[30]. DMEM with phenol red was added to the apical compartment, while pre-warmed PBS 1× without phenol red was added to the basolateral compartment for 2 h at 37 °C under 5% CO2 atmosphere. The optical density (OD) of the basolateral solution was measured at 450 nm using a spectrophotometer (Beckman, Pasadena, CA, USA) and only confluent Caco-2 polarized monolayers (characterized by a measure of OD at 450 nm of the basolateral medium less than 0.02) were used for co-culture experiments[31].
In vitro treatments
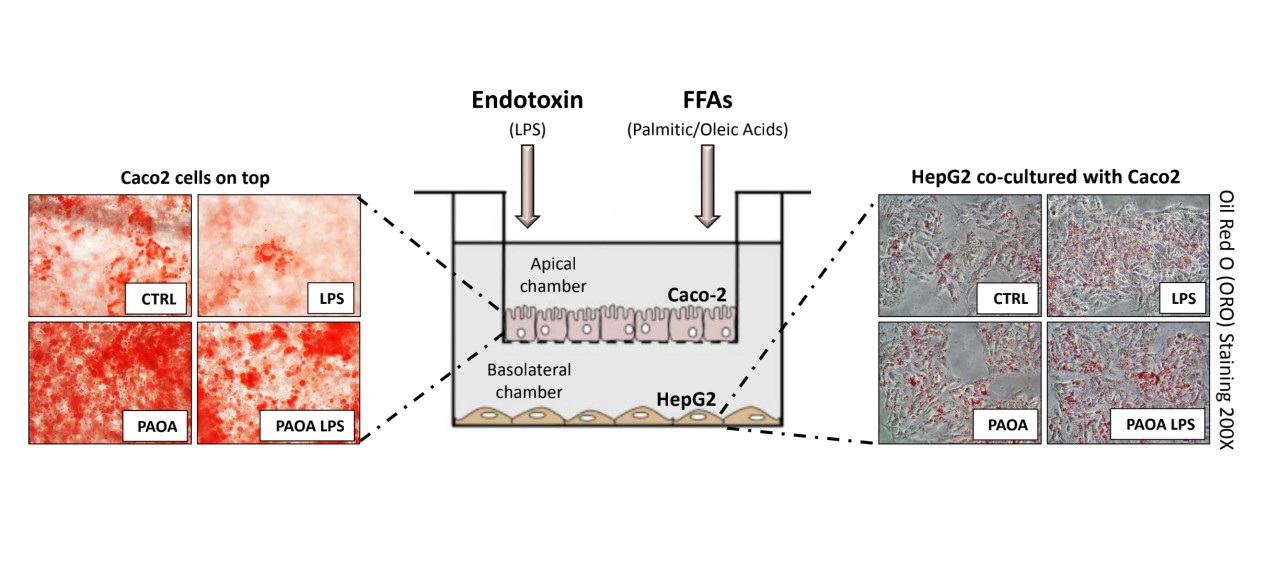
HepG2 cells were seeded at the concentration of 3.5 × 105 in multi-well plates and cultured for 48 h. Then, confluent monolayers of intestinal Caco-2 cells on Transwells were transferred on top of multi-well culture plates containing 90% confluent HepG2 cells, at the bottom. Caco-2 and HepG2 cells were incubated together in a co-culture system to recreate the bidirectional interplay between gut and liver. After an overnight quiescence in DMEM, 0.5% FBS, 100 ng/mL LPS (Sigma-Aldrich, Amsterdam, The Netherlands), and/or a mixture of palmitic and oleic acids at the final concentration of 0.25 mM (ratio 1:2, Sigma-Aldrich) were added in the quiescent medium of the apical chamber, whereas fresh quiescent medium was added to the basolateral compartment. After 24 h of incubation, co-cultured cells and apical and basolateral media were collected for further analyses. The optimal LPS and FFAs concentrations were determined by previous experiments[32]. A schematic representation of the Caco-2/HepG2 co-culture system is illustrated in Figure 1.

Figure 1. Schematic overview of the Caco-2/HepG2 co-culture system. Caco-2 cells were grown and differentiated on polycarbonate filter membranes (0.4 μm semipermeable pore diameter) for 21 days to recreate intestinal barrier. Then, Transwells were transferred on the top of confluent HepG2 cells seeded in multi-well plates and treated with endotoxin (LPS) and/or palmitic and oleic acids (FFAs) to mimic gut-liver interplay during NAFLD onset. NAFLD: Nonalcoholic fatty liver disease.
Epithelial permeability assay
To test the integrity of Caco-2 confluent monolayer, which mimics the intestinal barrier[33], in response to FFAs and/or LPS stimulation, we measured the transepithelial flux of 4 kDa fluorescein isothiocyanate (FITC)-labeled dextrans (FD4, Sigma Aldrich) as previously described[34,35]. Briefly, intestinal monolayers were equilibrated with pre-warmed PBS 1× at 37 °C, and then the fluorescent FD4 (1 mg/mL in PBS 1×) was added to the apical chamber. Then, 100 μL of sample were removed from the basolateral compartment at 1 h intervals for 4 h, and the volume removed was replaced with warmed PBS 1×. Epithelial permeability was compared between the different experimental conditions in quadruplicate, at the end of the 4 h. Fluorescence intensity (excitation 492 nm/emission 520 nm) was measured on a Promega GloMax plate reader. Concentrations of FD4 passed through the gut barrier were determined by linear regression using known dilutions of FITC-dextrans in PBS 1×.
Lipid secretion
Basolateral media of co-cultured cells were collected and concentrated using the Vivaspin 20/10 KDa polyethersulfone twin membrane (Sartorius Stedim Biotech, France), with the purpose to quantify apolipoprotein B (ApoB) release through enzyme linked immunosorbent assay (ELISA), which detects both ApoB main isoforms, the intestinal ApoB-48 and the hepatic ApoB-100, according to the manufacturer’s instructions (ThermoFisher Scientific). The lower limit of ApoB detection was 0.21 ng/mL.
The same methodological procedure was applied to determine TG-lipoprotein, TG, and FFA concentrations in basolateral media, by using sensitive enzyme-based methods (Abcam, Cambridge, UK).
All quantifications were normalized for the total protein content.
Cytokine secretion
Basolateral concentrated media were also exploited to quantify cytokine secretion through ELISA assays to detect TNF-α, IL6, and IL1β (R&D Systems, Minneapolis, USA). The lower limits of detection were 15.6, 3.9, and 9.4 pg/mL, respectively. Data were normalized to the total protein content.
Quantification of DNA damage
Total DNA was extracted from cell lysates of Caco2 and HepG2 by using DNeasy Blood & Tissue Kits (Qiagen, Hilden, Germany). Then, oxidative DNA damage induced by free radicals and other reactive species was measured as the formation of a-basic sites in genomic DNA, exploiting an aldehyde reactive probe that interacts with an aldehyde group on the open ring form of apurinic/apyrimidinic (AP) sites. The latter are common DNA lesions developed in the presence of enhanced oxidative stress. AP sites are tagged with biotin residues that can later be quantified using a streptavidin-enzyme conjugate, detected at
Gene expression analysis
RNA was extracted from cell cultures using Trizol reagent (ThermoFisher Scientific, Waltham, USA). Then, 1 μg of total RNA was retro-transcribed with VILO random hexamers synthesis system (ThermoFisher Scientific), and real-time quantitative reverse transcription PCR (qRT-PCR) was performed with a QuantStudio 3 Real-Time PCR thermocycler (ThermoFisher Scientific) using SYBR Green chemistry (Fast SYBR Green Master Mix; ThermoFisher Scientific) for all genes following the manufacturer’s instructions. All reactions were delivered in triplicate. Data were normalized for β-actin gene expression, and the results were expressed as arbitrary units or fold increase as indicated in violin plots. All primers are listed in Table 1.
Sequence of primers used in quantitative real-time PCR experiments
| Forward 5’→3’ | Reverse 5’→3’ | |
| ATF4 | AAACCTCATGGGTTCTCCAG | GGCATGGTTTCCAGGTCACT |
| ATF6 | AATTCTCAGCTGATGGCTGT | TGGAGGATCCTGGTGTCCAT |
| FASn | GCTGGGTGGAGTCTCTGAAG | TGCAACACCTTCTGCAGTTC |
| GRP78 | CTTGCCGTTCAAGGTGGTTG | CTGCCGTAGGCTCGTTGAT |
| MnSOD2 | CAAATTGCTGCTTGTCCAAA | TCTTGCTGGGATCATTAGGG |
| MTTP | GCTTCCGTTAAAGGTCACACA | CAGCTGTTATCGTGACTTGGA |
| PPARα | ATGGCATCCAGAACAAGGAG | TCCCGTCTTTGTTCATCACA |
| SREBP1 | TGCATTTTCTGACACGCTTC | CCAAGCTGTACAGGCTCTCC |
| TNF-α | TCAGCCTCTTCTCCTTCCTG | TGAGGTACAGGCCCTCTGAT |
| XBP1 | GAAGCCAAGGGGAATGAAGT | GCCCAACAGGATATCAGACTC |
| β-actin | GCTACAGCTTCACCACCACA | AAGGAAGGCTGGAAAAGAGC |
Western blot analysis
Total protein lysates were extracted from co-cultures using RIPA buffer containing phosphatase and protease inhibitors (PhosSTOP and cOmplete™ Protease Inhibitor Cocktails, Roche-Sigma Aldrich). Samples were pooled prior to electrophoretic separation and all reactions were performed in duplicate. Then, equal amounts of proteins (60 μg) were separated by SDS-PAGE, transferred electrophoretically to nitrocellulose membrane (BioRad, Hercules, CA), and incubated with specific antibodies overnight. At least three independent lots of freshly extracted proteins were used for experiments. The antibodies and concentrations used are listed in Table 2.
List of antibodies and relative dilutions used in Western blotting experiments
| Antibody | Catalog number |
| APOB (1:1000 WB) | ThermoFisher #HYB0690202 |
| Claudin 1 (1:1000 WB) | ThermoFisher #2H10D10 37-4900 |
| Claudin 2 (1:1000 WB) | ThermoFisher #12H12 32-5600 |
| p (Ser800)-TLR4 | ThermoFisher #PA5-105713 |
| TLR4 | ThermoFisher #48-2300 |
| ZO-1 (1:500 WB) | ThermoFisher #33-9100 |
| Vinculin (1:5000 WB) | Abcam # ab129002 |
| β-actin (1:5000 WB) | Abcam #ab6276 |
Oil red O staining
Oil red O (ORO) is a soluble red powder with high affinity for neutral triglycerides and lipids stored in the lipid droplets. In brief, co-cultures were gently washed twice with 1 mL of sterile PBS 1×, and then cells were fixed with 4% formalin for 15 min at room temperature. Next, each sample was washed with 1 mL of sterile water and 60% isopropyl alcohol was added for 5 min. ORO working solution was prepared by mixing three parts of ORO stock solution (3 mg/mL in isopropyl alcohol 100%) and two parts of sterile water, followed by filtration in a filter funnel. Next, 1 mL/well of ORO working solution was added to each sample and left for 45 min. Finally, plates were gently rinsed with tap water, paying attention not to disrupt monolayers, and lipid content was visualized in red color. ORO positive areas were quantified by ImageJ software (1.51a version) in 10 random non-overlapping micrographs (at magnification 200×) by calculating the ORO positive area as percentage of pixels above the threshold value with respect to the total pixels per area[32].
Statistical analyses
Statistical analyses were performed using JMP Pro 16.0 (SAS, Cary, NC) and Prism software (version 9.1.0, GraphPad Software) by using one-way analysis of variance (ANOVA) which was followed by post hoc t-tests adjusted for the number of comparisons when multiple groups were involved (Bonferroni correction). The results are shown as means ± SD or median and interquartile range, for violin plots, of at least three independent experiments, each containing triplicates or quadruplicates. P values lower than 0.05 (at two-tailed) were considered statistically significant (***P < 0.0001, **P < 0.001, *P < 0.01).
RESULTS
Caco-2/HepG2 co-culture system resembles the derangement of intestinal barrier integrity in presence of endotoxin and fatty acids
To investigate whether Caco-2 polarized monolayers are affected by endotoxin and/or fatty acids, we added in the quiescent medium of the apical chamber 100 ng/mL LPS and/or a combination of PAOA at the concentration of 0.25 mM, to recapitulate the clinical condition of NAFLD patients in which intestinal dysbiosis and excessive FFAs have been reported[10]. Then, to test the integrity of Caco-2 confluent monolayers, which recreate intestinal barrier[33], we measured the transepithelial flux of FD4. FD4 permeability across monolayers was increased after the exposure to LPS or PAOA, but more so in the presence of both (P < 0.0001, one-way ANOVA; adjusted P < 0.0001 for PAOA LPS vs. PAOA or LPS alone; Figure 2A). In keeping with the enhanced permeability, protein expressions of ZO1, claudin-1, and claudin-2, tight-junctions associated proteins, were also decreased (P < 0.0001, one-way ANOVA; adjusted P < 0.001 for PAOA LPS vs. PAOA or LPS alone; Figure 2B-D), thus mirroring what occurs in NAFLD patients, in which gut permeability is increased according to the disruption of intercellular tight junctions[12,38].

Figure 2. Intestinal permeability was further increased by the co-presence of LPA and PAOA. Polarized monolayers of Caco-2 cells co-cultured with HepG2 were assayed for paracellular permeability to 4 kDa FITC-dextrans (FD4). The amount of dextrans diffused from the apical to the basolateral compartments was determined by linear regression at 4 h using known standard concentrations of FD4. The results are represented as mean ± standard deviation of three independent experiments in triplicate (A). Tight junction-associated protein levels were evaluated in lysates from Caco-2 cells co-cultured with HepG2 in the presence or not of LPS and/or PAOA using Western blot: ZO-1 (B); Claudin 1 (C); and Claudin 2 (D). The expressions were normalized for β-actin as the housekeeping gene. For each condition, fresh protein lysates of at least three independent experiments were pooled. ***P < 0.0001 according to one-way ANOVA. LPA: lysophosphatidic acid; PAOA: palmitic and oleic acid; ANOVA: one-way analysis of variance; ZO-1: zonula occludens 1.
Caco-2/HepG2 co-cultures secrete TGs in response to FFAs and more so in the co-presence of LPS
We next examined whether Caco-2 monolayers exposed to PAOA and LPS accumulate lipid droplets by exploiting ORO staining to mark neutral lipid content. As expected, Caco-2 stimulated with PAOA displayed enhanced ORO positive areas and intracellular TG content [P < 0.0001, one-way ANOVA; adjusted P < 0.0001 for PAOA vs. untreated cells (Ctrl); Figure 3A-C]. However, the co-stimulation of PAOA with LPS did not further exacerbate fat deposition but rather favored TG disposal (adjusted P < 0.0001 for PAOA LPS vs. PAOA alone; Figure 3A-C). Consistently, LPS alone or combined with PAOA enforced TG dismissal, promoting ApoB [Figure 3D], TG-lipoprotein [Figure 3E], TG [Figure 3F], and FFA [Figure 3G] release in basolateral media (P = 0.002, P < 0.0001, P = 0.004, and P < 0.0001, one-way ANOVA, respectively; adjusted P < 0.05 for PAOA LPS vs. PAOA for all comparisons). These observations collectively suppor the notion that endotoxemia may boost the secretion of triglyceride-rich lipoproteins, fueling the creation of a pro-inflammatory context and resembling a condition named “lipemia of sepsis”[39,40].

Figure 3. Caco-2 polarized monolayers released TGs in response to FFAs and more so in the co-presence of LPS. Fat accumulation in Caco-2 polarized monolayers on top of the co-culture system was evaluated by ORO staining (200× magnification) and quantified by ImageJ software in 10 random non-overlapping micrographs for each experimental condition, by calculating the percentage of pixels above the threshold value with respect to the total pixels per area. At least three independent experiments were conducted (A, B). Intracellular TG content was assessed in lysates from Caco-2 polarized monolayers (C). Assessment of APOB (D), TG-rich lipoproteins (E), TGs (F) and FFAs (G) secreted were measured in concentrated culture media from the basolateral compartment and normalized for the total protein content. For violin plots, data were expressed as median concentration (thick dashed lines) and interquartile range (dotted lines). At least three independent experiments were conducted and pooled together. ***P < 0.0001, *P < 0.01 according to one-way ANOVA. TGs: Triglycerides; FFAs: free fatty acids; LPS: lipopolysaccharides; ORO: oil red O; APOB: apolipoprotein B; ANOVA: one-way analysis of variance.
HepG2 cells co-cultured with Caco-2 exposed to FFAs and LPS accumulated intracellular lipid droplets
As a consequence of the enhanced lipid secretion, HepG2 co-cultured with Caco-2 exposed to LPS, PAOA, or both accumulated lipid droplets and increased intracellular TG content (P < 0.0001, one-way ANOVA; adjusted P < 0.01 for PAOA LPS vs. PAOA alone; Figure 4A-C), as a result of lipid secretion from Caco-2 polarized monolayers. In this model, intracellular fat accumulation was paralleled by an induction of the lipogenic program, as detected by the conspicuous upregulation of sterol regulatory element binding protein 1-c and fatty acid synthase mRNA levels in HepG2 cells exposed to Caco-2 monolayers stimulated with PAOA and LPS (P < 0.0001, one-way ANOVA; adjusted P < 0.001 for PAOA LPS vs. PAOA alone for both comparisons; Figure 4D and E). On the other hand, the expression of genes regulating β-oxidation, i.e., peroxisome proliferator-activated receptor alpha, and very low-density lipoprotein (VLDL) export, i.e., microsomal triglyceride transfer protein, was suppressed, sustaining fat overload (P < 0.0001, one-way ANOVA; adjusted P < 0.001 for PAOA LPS vs. PAOA alone for both comparisons; Figure 4F and G). Superimposable results have previously been reported in patients and in preclinical models of NAFLD[41-43].

Figure 4. HepG2 cells co-cultured with Caco-2 exposed to FFAs and LPS accumulated intracellular lipid droplets. Fat accumulation in HepG2 on bottom of the co-culture system was evaluated by ORO staining (200× magnification) and quantified by ImageJ software in 10 random non-overlapping micrographs for each experimental condition by calculating the percentage of pixels above the threshold value with respect to the total number of pixels per area. At least three independent experiments were conducted (A, B). Intracellular TG content was assessed in lysates from HepG2 cells (C). (D, E) SREBP1c and FASn (genes involved in de novo lipogenesis); (F) PPARα (marker of β-oxidation); (G) and MTTP expressions were evaluated by qRT-PCR in HepG2 cell lysates; mRNA levels were normalized to β-actin. Data are expressed as fold increase compared to Ctrl. For violin plots, data are expressed as median concentration (thick dashed lines) and interquartile range (dotted lines). At least three independent experiments were conducted and pooled together. ***P < 0.0001 according to one-way ANOVA. FFAs: Free fatty acids; LPS: lipopolysaccharides; ORO: oil red O; SREBP1c: sterol regulatory element binding protein 1-c; FASn: fatty acid synthase; PPARα: peroxisome proliferator-activated receptor alpha; MTTP: microsomal triglyceride transfer protein; ANOVA: one-way analysis of variance.
Caco-2 polarized monolayers secreted pro-inflammatory cytokines in response to LPS, and the effect was amplified by the co-presence of FFAs
To ascertain the role of Caco-2 monolayers treated with PAOA and LPS in the promotion of a pro-inflammatory microenvironment, we quantified cytokine secretion in basolateral media. As expected,

Figure 5. Caco-2 polarized monolayers released pro-inflammatory cytokines in response to LPS and more so in the co-presence of FFAs. Secretion of TNF-α (A), IL6 (B), and IL1β (C) was measured in concentrated culture media from the basolateral compartment and normalized for the total protein content. pTLR4 and total TLR4 protein levels were evaluated in lysates from HepG2 cells co-cultured with Caco-2 stimulated with LPS and/or PAOA using Western blot. The expressions were normalized for the housekeeping gene β-actin. For each condition, fresh protein lysates of at least three independent experiments were pooled (D). TNF-α mRNA levels were evaluated by qRT-PCR in HepG2 cell lysates and normalized to β-actin (E). For violin plots, data are expressed as median concentration (thick dashed lines) and interquartile range (dotted lines). At least three independent experiments were conducted and pooled together. ***P < 0.0001, **P < 0.001 according to one-way ANOVA. LPS: Lipopolysaccharides; FFAs: free fatty acids; TNF-α: tumor necrosis factor alpha; IL6: interleukin 6; TLR4: toll-like receptor 4; PAOA: palmitic and oleic acid; ANOVA: one-way analysis of variance.
Caco-2/HepG2 co-cultures remodeled the switching from simple steatosis to NASH
We next assessed the impact of TG accumulation and inflammation driven by intestinal barrier derangement on the development of endoplasmic (ER) and oxidative stress, which are the hallmarks of NASH[36,45]. We demonstrated that HepG2 co-cultured with Caco-2 exposed to a combination of PAOA and LPS strongly upregulated the expression of markers of ER stress and unfolded protein response, including activating transcription factor 4 (ATF4), ATF6, X-box binding protein 1, and GRP78 (P = 0.0006 for ATF4 and P < 0.0001, one-way ANOVA for all genes; adjusted P < 0.05 for PAOA LPS vs. PAOA alone for all comparisons; Figure 6A-D). Oxidative stress was also triggered in HepG2 co-cultured with Caco-2 exposed to a combination of PAOA and LPS, as testified by the elevation of ROS/reactive nitrite species (RNS) production, H2O2, aldehyde derivate [malondialdehyde (MDA)] concentrations, and apurinic/apyrimidinic sites, the main ROS-induced DNA damage (P < 0.0001, one-way ANOVA; adjusted P < 0.01 for PAOA LPS vs. PAOA alone for all comparisons; Figure 6E-H). Similarly, the expression of the superoxide dismutase (MnSOD2) was further increased in HepG2 exposed to Caco-2 monolayers treated with PAOA and LPS, possibly as a compensatory mechanism to dampen ROS overflowing (P < 0.0001, one-way ANOVA; adjusted P < 0.01 for PAOA LPS vs. PAOA; Figure 6I).

Figure 6. HepG2 cells co-cultured with Caco-2 exposed to FFAs and LPS displayed an enhanced ER and oxidative stress. The expressions of ATF4 (A), ATF6 (B), XBP1 (C), and GRP78 (D), markers of ER stress were evaluated by qRT-PCR in HepG2 cell lysates and normalized using β-actin as the housekeeping gene. Data are expressed as fold increase compared to Ctrl. ROS/RNS content was measured using a colorimetric assay (Abcam) (E). H2O2 levels were quantified in HepG2 lysates through DCF ROS/RNS Colorimetric Assay Kit (Abcam) (F). Malondialdehyde (MDA) was colorimetrically measured in HepG2 lysates following the manufacturer’s instructions (Abcam) (G). The number of apurinic/apyrimidinic (AP) sites was obtained by isolating total DNA from HepG2 lysates and then it was normalized to the total amount of DNA extracted (H). MnSOD2 mRNA levels were assessed by qRT-PCR and normalized to β-actin (I). For violin plots, data are expressed as median concentration (thick dashed lines) and interquartile range (dotted lines). At least three independent experiments were conducted and pooled together. ***P < 0.0001, **P < 0.001 according to one-way ANOVA. FFAs: Free fatty acids; LPS: lipopolysaccharides; ER: endoplasmic reticulum; ATF4: activating transcription factor 4; ATF6: activating transcription factor 6; XBP1: X-box binding protein 1; ROS: reactive oxygen species; RNS: reactive nitrite species; ANOVA: one-way analysis of variance.
Superimposable findings have been obtained in basolateral media, in which harmful reactive species are elevated as a result of the exposure to PAOA and/or LPS (P < 0.0001, one-way ANOVA; adjusted P < 0.01 for PAOA LPS vs. PAOA alone for all comparisons; Supplementary Figure 1A-C). We cannot rule out that these triggers released from both damaged cells may exert a direct detrimental effect on gut barrier, further precipitating the derangement of intestinal tight-junctions integrity.
DISCUSSION
In this study, we examined the impact of endotoxins and FFAs on the alteration of the intestinal barrier integrity and fat accumulation in hepatocytes, which frequently co-occur in severe NAFLD, by using an in vitro model of Caco-2/HepG2 co-culture, in an attempt to mirror the gut-liver axis.
The human intestinal Caco-2 cell line was isolated, for the first time, by colon adenocarcinoma by
HepG2 cells, derived from human hepatocellular carcinoma, are usually used in metabolic studies, since they retain cell differentiation that is lost by primary hepatocytes in culture, preserving their ability to secrete plasma lipoproteins, internalize chylomicrons, regulate cholesterol metabolism and bile acid synthesis, and express inducible cytochrome P450 pathway[48].
Here, we combined the use of these two different cell types to reproduce as far as possible the gut-liver crosstalk, which occurs in vivo. Our results demonstrate that differentiated Caco-2 monolayers are susceptible to the apical exposure to noxious concentrations of a combination of PAOA and LPS, significantly altering FD4 permeability and tight-junction integrity. Our observations are aligned with the current concept that Western diet and high-fat diet administration in rodents disrupted gut barrier integrity and induced pro-inflammatory luminal LPS transition into the blood flow, contributing to liver damage[13,14,49]. Moreover, the incubation of HepG2 cultures with effectors released in basolateral media from Caco-2 boosts fat deposition, paralleled by TLR4 activation and severe oxidative damage, resulting in lipid peroxidation and ROS-triggered DNA injuries. Our findings confirm that elevated concentration of serum LPS forces the activation of TLR4/NF-κB signaling in hepatocytes, thus driving, in turn, pro-inflammatory cytokine secretion and ROS production.
The relevance of gut-liver interplay in NAFLD susceptibility has been broadly addressed in both preclinical and clinical studies, although it remains a matter of debate. Despite the relatively standard co-culture conditions that we chose for the growth and maintenance of the Caco-2 and HepG2 cell lines, this is the first study, to our knowledge, that adopts this system to resemble human NAFLD and deeply investigate the mechanisms underlying NASH development. Our co-culture method is surely technically limited compared to novel and more advanced three-dimensional cell culture protocols (i.e., spheroids, organoids, 3D bio-printed tissues, and many others), however it seems to be fully reproducible, cost-effective, and capable of mirroring the interactions between the intestinal tract and the liver during the natural history of human disease.
CONCLUSION
The Caco-2/HepG2 co-culture system may faithfully reproduce the breach in the intestinal barrier integrity that occurs in NAFLD, thus resulting in the increased inflammatory response and oxidative and ER stress, which in turn promote the switch towards NASH. This study paves the way for future investigations, exploiting co-culture platforms to pinpoint precise cell-to-cell communications and soluble mediators exchanged between different cell types during progressive NAFLD onset and to test the efficacy of novel therapeutic strategies or dietary approaches to this disease.
DECLARATIONS
Authors’ contributionsWriting the manuscript, preparing figures, read and approved the final draft: Meroni M, Paolini E, Longo M, Piciotti R, Tria G, Fargion S, Fracanzani AL, Dongiovanni P
All authors have read and agreed with the published version of the manuscript.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe study was supported by Ricerca Corrente Fondazione IRCCS Cà Granda and Ricerca Finalizzata Ministero della Salute GR-2019-12370172.
Conflicts of interestAll authors declared there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
Supplementary MaterialsREFERENCES
1. Meroni M, Longo M, Paolini E, et al. MAFLD definition underestimates the risk to develop HCC in genetically predisposed patients. J Intern Med ;2021:374-6.
2. Younossi Z, Henry L. Contribution of alcoholic and nonalcoholic fatty liver disease to the burden of liver-related morbidity and mortality. Gastroenterology 2016;150:1778-85.
3. Eslam M, Sanyal AJ, George J. International Consensus Panel. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020;158:1999-2014.e1.
4. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73-84.
5. Wong RJ, Aguilar M, Cheung R, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015;148:547-55.
6. Dongiovanni P, Meroni M, Longo M, Fargion S, Fracanzani AL. Genetics, immunity and nutrition boost the switching from NASH to HCC. Biomedicines 2021;9:1524.
7. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 2010;52:1836-46.
8. Meroni M, Longo M, Dongiovanni P. Alcohol or gut microbiota: who is the guilty? Int J Mol Sci 2019;20:4568.
9. Poeta M, Pierri L, Vajro P. Gut-liver axis derangement in non-alcoholic fatty liver disease. Children (Basel) 2017;4:66.
10. Meroni M, Longo M, Dongiovanni P. The role of probiotics in nonalcoholic fatty liver disease: a new insight into therapeutic strategies. Nutrients 2019;11:2642.
11. Giorgio V, Miele L, Principessa L, et al. Intestinal permeability is increased in children with non-alcoholic fatty liver disease, and correlates with liver disease severity. Dig Liver Dis 2014;46:556-60.
12. Miele L, Valenza V, La Torre G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009;49:1877-87.
13. Lindstedt G, Lindstedt S, Gustafsson BE. Mucus in intestinal contents of germfree rats. J Exp Med 1965;121:201-13.
14. Wang L, Fouts DE, Stärkel P, et al. Intestinal REG3 lectins protect against alcoholic steatohepatitis by reducing mucosa-associated microbiota and preventing bacterial translocation. Cell Host Microbe 2016;19:227-39.
15. Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 2012;3:279-88.
16. Boulangé CL, Neves AL, Chilloux J, Nicholson JK, Dumas ME. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med 2016;8:42.
17. Del Chierico F, Nobili V, Vernocchi P, et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017;65:451-64.
18. Delarue J, Lallès JP. Nonalcoholic fatty liver disease: roles of the gut and the liver and metabolic modulation by some dietary factors and especially long-chain n-3 PUFA. Mol Nutr Food Res 2016;60:147-59.
19. Bibbò S, Ianiro G, Dore MP, Simonelli C, Newton EE, Cammarota G. Gut microbiota as a driver of inflammation in nonalcoholic fatty liver disease. Mediators Inflamm 2018;2018:9321643.
20. Puri P, Sanyal AJ. The intestinal microbiome in nonalcoholic fatty liver disease. Clin Liver Dis 2018;22:121-32.
21. Borrelli A, Bonelli P, Tuccillo FM, et al. Role of gut microbiota and oxidative stress in the progression of non-alcoholic fatty liver disease to hepatocarcinoma: current and innovative therapeutic approaches. Redox Biol 2018;15:467-79.
22. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016;65:1038-48.
23. Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 2014;146:1513-24.
24. Ferreira DF, Fiamoncini J, Prist IH, Ariga SK, de Souza HP, de Lima TM. Novel role of TLR4 in NAFLD development: modulation of metabolic enzymes expression. Biochim Biophys Acta 2015;1851:1353-9.
25. Cai C, Zhu X, Li P, et al. NLRP3 deletion inhibits the non-alcoholic steatohepatitis development and inflammation in Kupffer cells induced by palmitic acid. Inflammation 2017;40:1875-83.
26. Ray K. NAFLD. Leaky guts: intestinal permeability and NASH. Nat Rev Gastroenterol Hepatol 2015;12:123.
27. Meroni M, Longo M, Rametta R, Dongiovanni P. Genetic and epigenetic modifiers of alcoholic liver disease. Int J Mol Sci 2018;19:3857.
28. De Gregorio V, Telesco M, Corrado B, et al. Intestine-liver axis on-chip reveals the intestinal protective role on hepatic damage by emulating ethanol first-pass metabolism. Front Bioeng Biotechnol 2020;8:163.
29. Breemen RB, Li Y. Caco-2 cell permeability assays to measure drug absorption. Expert Opin Drug Metab Toxicol 2005;1:175-85.
30. Garcia MN, Flowers C, Cook JD. The Caco-2 cell culture system can be used as a model to study food iron availability. J Nutr 1996;126:251-8.
31. Tayyeb JZ, Popeijus HE, Mensink RP, Plat J. Butyric acid added apically to intestinal Caco-2 cells elevates hepatic ApoA-I transcription and rescues lower ApoA-I expression in inflamed HepG2 cells co-cultured in the basolateral compartment. Biomolecules 2021;11:71.
32. Meroni M, Dongiovanni P, Longo M, et al. Mboat7 down-regulation by hyper-insulinemia induces fat accumulation in hepatocytes. EBioMedicine 2020;52:102658.
33. Sambuy Y, De Angelis I, Ranaldi G, Scarino ML, Stammati A, Zucco F. The Caco-2 cell line as a model of the intestinal barrier: influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol Toxicol 2005;21:1-26.
34. Twarog C, Liu K, O'Brien PJ, et al. A head-to-head Caco-2 assay comparison of the mechanisms of action of the intestinal permeation enhancers: SNAC and sodium caprate (C10). Eur J Pharm Biopharm 2020;152:95-107.
35. He S, Liu F, Xu L, et al. Protective effects of ferulic acid against heat stress-induced intestinal epithelial barrier dysfunction in vitro and in vivo. PLoS One 2016;11:e0145236.
36. Longo M, Meroni M, Paolini E, et al. TM6SF2/PNPLA3/MBOAT7 loss-of-function genetic variants impact on NAFLD development and progression both in patients and in in vitro models. Cell Mol Gastroenterol Hepatol 2021;13:759-88.
37. Longo M, Paolini E, Meroni M, et al. α-lipoic acid improves hepatic metabolic dysfunctions in acute intermittent porphyria: a proof-of-concept study. Diagnostics (Basel) 2021;11:1628.
38. Kessoku T, Kobayashi T, Tanaka K, et al. The role of leaky gut in nonalcoholic fatty liver disease: a novel therapeutic target. Int J Mol Sci 2021;22:8161.
39. Barcia AM, Harris HW. Triglyceride-rich lipoproteins as agents of innate immunity. Clin Infect Dis 2005;41 Suppl 7:S498-503.
40. Harris HW, Gosnell JE, Kumwenda ZL. The lipemia of sepsis: triglyceride-rich lipoproteins as agents of innate immunity. J Endotoxin Res 2000;6:421-30.
41. Meroni M, Longo M, Erconi V, et al. mir-101-3p downregulation promotes fibrogenesis by facilitating hepatic stellate cell transdifferentiation during insulin resistance. Nutrients 2019;11:2597.
42. Dongiovanni P, Meroni M, Baselli GA, et al. Insulin resistance promotes Lysyl Oxidase Like 2 induction and fibrosis accumulation in non-alcoholic fatty liver disease. Clin Sci (Lond) 2017;131:1301-15.
44. Ceccarelli S, Panera N, Mina M, et al. LPS-induced TNF-α factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget 2015;6:41434-52.
45. Ashraf NU, Sheikh TA. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free Radic Res 2015;49:1405-18.
46. Fogh J, Fogh JM, Orfeo T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst 1977;59:221-6.
47. Ferruzza S, Rossi C, Scarino ML, Sambuy Y. A protocol for differentiation of human intestinal Caco-2 cells in asymmetric serum-containing medium. Toxicol In Vitro 2012;26:1252-5.
48. Javitt NB. Hep G2 cells as a resource for metabolic studies: lipoprotein, cholesterol, and bile acids. FASEB J 1990;4:161-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Meroni M, Paolini E, Longo M, Piciotti R, Tria G, Fargion S, Fracanzani AL, Dongiovanni P. Recreating gut-liver axis during NAFLD onset by using a Caco-2/HepG2 co-culture system. Metab Target Organ Damage 2022;2:4. http://dx.doi.org/10.20517/mtod.2021.19
AMA Style
Meroni M, Paolini E, Longo M, Piciotti R, Tria G, Fargion S, Fracanzani AL, Dongiovanni P. Recreating gut-liver axis during NAFLD onset by using a Caco-2/HepG2 co-culture system. Metabolism and Target Organ Damage. 2022; 2(2): 4. http://dx.doi.org/10.20517/mtod.2021.19
Chicago/Turabian Style
Meroni, Marica, Erika Paolini, Miriam Longo, Roberto Piciotti, Giada Tria, Silvia Fargion, Anna Ludovica Fracanzani, Paola Dongiovanni. 2022. "Recreating gut-liver axis during NAFLD onset by using a Caco-2/HepG2 co-culture system" Metabolism and Target Organ Damage. 2, no.2: 4. http://dx.doi.org/10.20517/mtod.2021.19
ACS Style
Meroni, M.; Paolini E.; Longo M.; Piciotti R.; Tria G.; Fargion S.; Fracanzani AL.; Dongiovanni P. Recreating gut-liver axis during NAFLD onset by using a Caco-2/HepG2 co-culture system. Metab Target Organ Damage. 2022, 2, 4. http://dx.doi.org/10.20517/mtod.2021.19
About This Article
Copyright

Data & Comments
Data


 Cite This Article 32 clicks
Cite This Article 32 clicks

 Like This Article 31
likes
Like This Article 31
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.