
Liver-derived lipoproteins and inflammation: from pathophysiology to pharmacological targets in metabolic liver disease
 0
0
Abstract
Low density lipoproteins (LDL) reduction remains the key goal for reducing the risk of atherosclerotic cardiovascular diseases (CVD) in people with high residual risk and metabolic complications including liver disease. Notwithstanding, epidemiological projections support a key role of liver-derived apolipoprotein B (ApoB) containing lipoproteins, namely very low density lipoproteins (VLDL) and their “remnants” (TG), undergoing the activity of lipases, in eliciting atherosclerotic inflammatory sequelae of a comparable order of magnitude to that of LDL. Disparate experimental evidence supports that triglycerides (TG), residual cholesterol content, or the large apolipoprotein set on the surface of these lipoproteins can elicit a number of plausible immune-inflammatory mechanisms that foster the vascular atherosclerotic process. Therapeutic options that convincingly lowered the plasma levels of liver-derived ApoB containing lipoproteins, either by reducing the hepatic synthesis or by improving the peripheral lipolysis of the lipid content, did not exert robust CVD risk reduction, and the effect on inflammation was questionable. Understanding the mechanisms linking liver-derived lipoproteins with chronic inflammation will provide pathophysiological insights for the identification of new therapeutic targets for people at high CVD risk and with metabolic complications. In this perspective, this topic is of immediate interest for the prevention of CVD in patients affected by non-alcoholic fatty liver disease (NAFLD) and, even more, for NAFLD patients with diabetes, insulin resistance, or other comorbidities (metabolic-associated fatty liver disease). This review resumes the principal physio-pathological insights regarding the metabolism of liver-derived lipoproteins and provides an update on the current pharmacological options that can be considered for improving CVD prevention in metabolic liver diseases.
Keywords
BACKGROUND
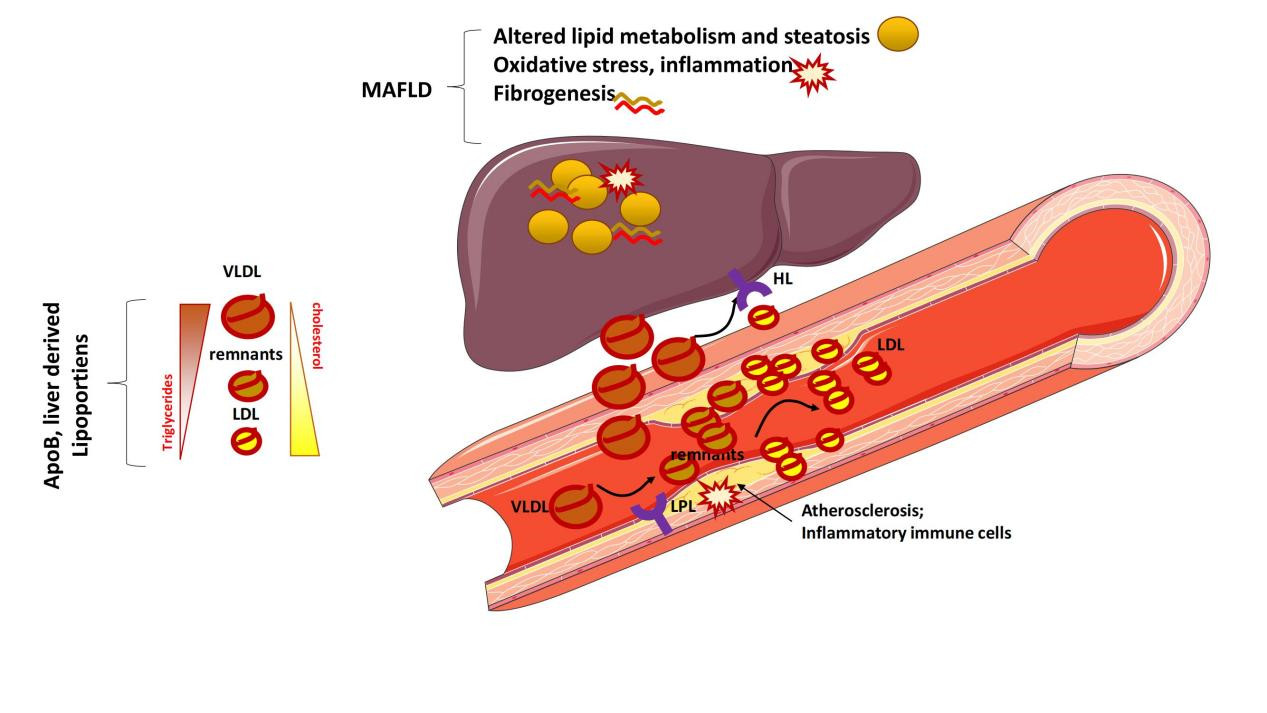
Non-alcoholic fatty liver disease (NAFLD) is a prevalent disease that increases the risk of cardiovascular disease (CVD) as compared to other liver diseases (e.g., infective liver disease)[1-3]. In the presence of systemic metabolic complications [i.e., metabolic-associated fatty liver disease (MAFLD)[4]], the CVD risk is even higher. MAFLD has predominate causes from nutrition overload to altered systemic metabolism and inflammation[3,4], together with persistent liver damage that eventually lead to the development of liver fibrosis and cirrhosis.
Mechanistically, the development of MAFLD encompasses complex molecular aspects which intertwine in pathological processes starting from alteration in lipid metabolism and pro-inflammatory activation[5]. All together, these mechanisms foster oxidative stress, cell apoptosis, and extracellular matrix formation up to the fibrogenesis process.
Alterations in lipid metabolism, defined as over-production of triglyceride-rich liver-derived lipoproteins [very low density lipoproteins (VLDL)], physiologically occurs in humans following consumption of a high-fat based meal (postprandial lipemia), but, in people with metabolic complications (including MAFLD), this is dramatically exacerbated[6-8]. This iterative process over time promotes a constantly elevated amount of VLDL, which over-engages the activity of peripheral lipases [lipoprotein lipase (LPL) and hepatic lipase (HL)], which are in charge of the lipolysis of the triglyceride (TG) content of VLDL[8].
Hence, the accumulation of VLDL will turn into a relative increase in the amount of remnant cholesterol in the downstream products of the VLDL, which are low density lipoproteins (LDL).
Mechanistically, both VLDL and LDL separately, which share apolipoprotein B (ApoB) on their surface, exert inflammatory and potent pro-atherogenic processes, which are discussed in this review. In response to the wealth of evidence from pre-clinical studies, the reduction of LDL is the first goal effectively outreached for reducing the risk of atherosclerotic CVD in people with high residual risk. By contrast, the pharmacological strategies thus far available are unable to provide a comparable magnitude of VLDL reduction[9,10].
Hence, the understanding of the alterations in lipid metabolism as a primum movens for these pathological sequelae is of immediate interest for NAFLD/MAFLD, by contrast to other types of liver disease. In fact, an in-depth study of these aspects might help to pave the road towards the development of future strategies controlling the over-production of VLDL and to more effectively reduce the entire set of atherogenic ApoB-containing liver-derived lipoproteins.
In this review, the concept of liver-derived lipoproteins and its relevance in inflammation and CVD are described and critically analyzed. Subsequently, this review summarizes in detail the pharmacological strategies and pipeline that are being tested and currently under development for future consideration in the prevention of CVD in metabolic liver disease.
THE METABOLISM OF LIPOPROTEINS DURING THE POSTPRANDIAL ENERGETIC CHARGE
Evolution developed the mechanism by which our body stores energy following consumption of a high fat meal and ensures a proper disposal in the case of long-term starvation. TGs are the principal forms of lipids that ensure an elevated potential energy per molecule as, when they are mobilized by LPL from visceral adipose tissue, they are fuel for high-energy-producing oxidative metabolism in high energy-demanding metabolic tissues, including the heart and skeletal muscle. Lipoproteins are key drivers of TG mobilization as, by virtue of their lipophilic nature, they cannot freely circulate in blood. Chylomicrons are the intestinally derived lipoproteins that increase in quantity immediately following the ingestion and absorption through the duodenal villi of the lipid content of a meal (no longer than 1 h[11]). Following arrival to the liver, the lipid material is immediately distributed to hepatocytes, which subsequently prepares this lipid material into another lipoprotein structure, i.e., VLDL. VLDLs are produced in a higher order of magnitude as compared to chylomicrons and, per particle, carry a TG as well as a proportion of cholesterol, which derives from the hepatic production pool. Both chylomicrons and VLDL undergo the activity of endothelium-bound lipases (both LPL and HL) to hydrolyze their triglyceride (TG) content, with VLDL being a preferential substrate of LPL and HL. A fine tuning of this enzymatic flow is essential to ensure the distribution of lipid energy sources to tissues. In fact, the activity of LPL and HL residing in visceral adipose tissue and the liver is enhanced following ingestion of a high fat meal, while it decreases in other oxidative tissues (e.g., skeletal muscle); conversely, in the fasting state, this ratio is the opposite, favoring the oxidative utilization and energy expenditure. These energetic flows among these metabolic sites are iterative over time, since the elevation of VLDL occurs in the so-called postprandial lipemia (PPL[12]), a physiological situation in which people from western societies spend the majority of their daily life, according to epidemiological projections. In fact, PPL has been recently described to last 6-8 h[13] following the consumption of a high fat meal (20-40 g of fats/meal) in affluent societies[14,15].
VLDL, although with a lower relative amount of TG as compared to chylomicrons, represents the predominant mediator of the energetic exchanges during PPL and over time, being higher in number as compared to chylomicrons and the preferential substrate of LPL. In addition, the iterative postprandial situation increases VLDL (which stays in circulation for 4-13 h on average)[16] [Figure 1]. As soon as VLDL undergoes the activity of lipases, it becomes smaller in size (from 30-70 to 20-25 nm in diameter) and with a higher residual proportion of remnant cholesterol (from 15% to 65% per particle)[16] [Figure 1]. This passage promotes the increase of lipoprotein density and makes the original VLDL remnant intermediate lipoproteins, and then low density lipoproteins (LDL). In the long term, the much higher half-life of LDL than that of VLDL (on average, 2.5-3.5 days for LDL [Figure 1]) explains the greater abundance of LDL particles, estimated at around 3-10 LDL per each VLDL in most individuals[16]. The quantity of cholesterol carried by these lipoproteins is much higher compared to that coming from the intestine. In fact, out of
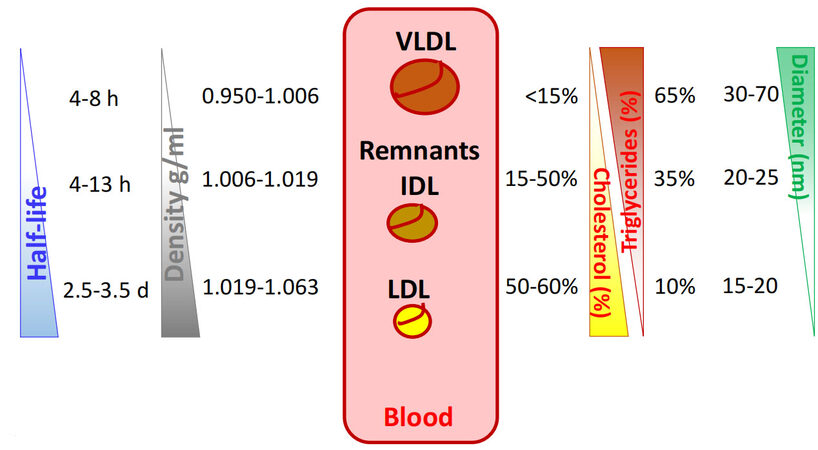
Figure 1. Half-life, density, cholesterol, triglyceride relative content, and diameter of liver-derived lipoproteins. This figure summarizes the ranges (average) of (from left to right) half-life, density, cholesterol, triglycerides relative content (percentage), and diameter of VLDL, Remnant/IDL, and LDL. ApoB: Apolipoprotein B; VLDL: very low density lipoproteins; IDL: intermediate density lipoproteins; LDL: low density lipoproteins.
LIVER-DERIVED LIPOPROTEINS AS POTENT CARDIOVASCULAR RISK PREDICTORS IN CLINIC
The clinical evidence, as does the epidemiological projection, supports that it is this exchange of cholesterol among lipoproteins that matters in the development of the atherosclerotic process. In fact, hyperchylomicronemia (the condition of elevated triglyceride rich, intestinally-derived chylomicrons in fasting and during PPL), as with severe hypertriglyceridemia (TG over 880 mg/dL), results in pancreatitis, metabolic complications, and liver alterations, but it does not increase CVD risk[20-22]. Mechanistically, in fact, large diameter chylomicrons (> 80 nm) and VLDL of immediate liver secretion (60-80 nm) are not able to transmigrate over the vascular endothelial layer. Conversely, CVD risk triples as TG increases from 250 to 450 mg/dL, indicating that only VLDL remnants and cholesterol-enriched LDL, the entire set of apolipoprotein B (ApoB)-containing lipoproteins with less TG content and with diameter less than 60 nm[23,24], can penetrate in the endothelial layer.
ApoB, by reflecting both the acute raise of triglyceride-rich ApoB-containing lipoproteins during PPL and the chronic accumulation of cholesterol, represents the key target of clinical situations associated with high CVD risk and characterized by elevated production of liver-derived lipoproteins.
ApoB is the protein structure present in equimolar ratio per each lipoprotein in VLDL, LDL, and all remnants, and it is in charge of interacting with receptors and the internalization of lipoproteins in peripheral tissues[25-27]. Statins, which inhibit the synthesis of cholesterol in the liver, lower LDL-C more than non-HDL-C and relatively more than the molar quantity of ApoB[28], indicating that reduction of cholesterol is not sufficient to control the hepatic secretion of all the ApoB-containing lipoproteins. Elegant Mendelian randomization studies that mimic the effects of CETP inhibitors and statins, by combining variants in the cholesterol ester transfer protein (CETP, in charge of exchanging cholesterol between VLDL and high density lipoproteins) and the 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR, the rate limiting step enzyme for cholesterol synthesis) genes (to create genetic scores) robustly affirmed the relevance in targeting ApoB, as a unique direct marker of both LDL and liver-derived VLDL and remnants[29]. In fact, a CETP score at or above the median was associated with lower levels of LDL-C, lower ApoB, and consistently lower CVD risk. This effect was similarly exerted by an HMGCR score at or above the median, which was associated with lower levels of LDL-C, ApoB, and CVD risk as well. However, the effect of both scores together was additive for LDL-C but not for ApoB or CVD risk. Indeed, the reduction of LDL-C in people harboring both scores equated to the sum of each independent score, although the extent of the reduction in ApoB and CVD risk were attenuated in people harboring both scores vs. those achieved by a single score[29]. Thus, Mendelian randomization indicates that the primary mechanism of benefit from lowering LDL-C relates to the lowering of the number of LDL particles, i.e., to the lowering of ApoB. Hence, ApoB unifies, amplifies, and simplifies the information from the conventional LDL-C lipid marker as to the atherogenic risk attributable to the liver-derived lipoproteins.
Familial combined hyperlipidemia (FCHL), a polygenic situation of elevated liver-derived ApoB particle production, reduced TG hydrolysis, resulting in liver steatosis, metabolic complications, and elevated CVD risk. Conversely, familial combined hypolipidemia (FHBL2), a rare genetic condition (OMIM#605019) driven by deficiency of angiopoietin-like 3 (Angptl3), a natural inhibitor of peripheral lipases, results in an improved postprandial response to fatty meal, minimal liver-derived lipoprotein production, and neutralized CVD risk[30,31]. Similarly, non-alcoholic fatty liver disease (NAFLD), where PPL increases because of elevated quantity of liver-derived VLDL, results in elevated susceptibility to CVD[1,2], which ranks as the first comorbidity, even before that of extrahepatic malignancies and liver-related complications[32,33]. Furthermore, when systemic metabolic complications are also present (i.e., MAFLD)[34], the peripheral hydrolysis of TG in VLDL by lipases is also reduced[6-8], further increasing CVD risk. The hardwired pathophysiological connections in MAFLD, however, complicate the understanding of the metabolism of lipoproteins as the primum movens. Indeed, the excess caloric intake is the first mover in promoting the systemic inflammation associated with metabolic complications and insulin resistance. In this scenario, liver-derived lipoproteins might act as both mediators of the link caloric excess-inflammation and simply an epiphenomenon of altered handling of the lipid energetic sources in liver and adipose tissue.
THE INFLAMMATORY POTENTIAL OF LIVER-DERIVED LIPOPROTEINS
Around 75 years ago, Mereton wrote that “the lipid particles must be assumed to be retained and deposited from the plasma-derived nutrient lymph stream which normally passes from the lumen through the intramural structures towards the adventitial venules and lymphatics. It may be theorised that the increased particle size of the lipids in sustained or alimentary hyperlipemia is the stimulus to the phagocytosis in the intima by macrophages and the formation of the typical foam cells”[35,36].
This pioneering concept anticipated the subsequent data indicating that ApoB containing VLDL is surveilled by immune-inflammatory checkpoints, and, by entering the sub-endothelial layer of vasculature, they directly contribute to the inflammatory mechanisms, including cholesterol deposition and pro-thrombotic effects[37-42], involved in the progression of atherosclerosis. Within these mechanisms, however, the experimental evidence produced thus far still questions whether it is the ApoB lipoprotein per se, or it is more likely their content of the aryl carbon chains of TG (fatty acids) or that of cholesterol[43].
Mechanisms elicited by TG in liver-derived lipoproteins
Fatty acids in lipoproteins can be medium chain (6-12 carbons) and long chain (up to 22 carbons) and can be saturated (SFA), monounsaturated (MUFA), or polyunsaturated (PUFA). Among PUFAs, those with a first double bond on the third carbon are referred to as n-3, whereas those with a first double bond on the sixth carbon are called n-6.
SFAs stimulate the inflammatory activation of macrophages by a process that involves toll-like receptor 4 (TLR4), a pattern recognition receptor that plays a key role in the innate patrolling of bacterial pathogens, including lipopolysaccharide (LPS). In fact, the activation of TLR4 by SFA induces an over-activation of
Mechanisms elicited by cholesterol in liver-derived lipoproteins
Cholesterol can be oxidized into different types of oxysterols by a number of cardiovascular risk determinants as well as by factors[56,57]. Oxysterols contribute to the formation of modified LDLs [namely, oxidized LDL (oxLDL)], which are taken up by macrophages in the atheroma. Within cells, the crystallization of excess cholesterol occurs, further increasing its atherogenic potential and the ability to evoke the inflammatory activation of effector inflammatory lymphocytes[58] and the induction of the inflammasome (NOD-like receptor protein 3) complex[59,60]. Acute exposure of macrophages to oxLDL prolongs these mechanisms by inducing epigenetic priming of a complex set of inflammatory players[61]. In addition, NLRP3 undergoes this epigenetic long-lasting activation, a process that has been described to favor an inflammatory phenotype of the myeloid hematopoietic immune compartment[62].
The apolipoprotein content of liver-derived lipoproteins
By in vivo[18] fluoro-deoxyglucose position emission tomography (PET) blood labeling to track metabolic circulating leukocytes during vascular inflammation, it was demonstrated that elevated hepatic VLDL and their remnants (such as in the case of FCHL), despite lower levels of LDL-C, elicit arterial inflammation compared with subjects with familial hypercholesterolemia (a genetic condition of unique elevated
These clinical observations recapitulate findings ex vivo as, per particle basis, cholesterol-rich liver-derived lipoproteins are more potent inducers of macrophage inflammatory foam cells than LDL alone and do not need structural modification to trigger uptake[66-68].
Human endothelial cells stimulated in vitro with fasting VLDL (concentration of 50 µg/mL in ApoB) isolated from patients with hypertriglyceridemia showed an exaggerated expression pattern of multiple inflammatory and adhesion molecule (VCAM-1 and PECAM-1). These mechanisms are even further induced by stimulation of these cells with VLDL for 4 h during PPL[69]. At the same time point, VLDL induced in vivo an increased number of circulating leukocytes[69], intracellular lipid accumulation[70], and cell activation, leading to adhesion to endothelium, thus suggesting that the endothelial activation during PPL is associated with immune response switching[70,71].
Furthermore, the inflammatory potential of liver-derived lipoproteins is supported by elegant pre-clinical studies showing that improving their catabolism (by improving the expression of their hepatic receptors via gene therapy approaches) would result in a significant regulation of the systemic inflammation, thus an improvement of metabolic fitness and cardioprotection[72].
In addition to ApoB, a large set of apolipoproteins characterize the membrane of liver-derived lipoproteins. In fact, the proteome of liver-derived lipoproteins includes other apolipoproteins (smaller-sized as compared to ApoB), including ApoCIII, ApoCII, ApoCI, ApoAIV, ApoAV, and ApoE, as well as regulators of the LPL activity, including ANGPTL-3, -4, and -8.
Of these, ApoE is a physiological mediator of the uptake of liver-derived lipoproteins by peripheral tissues and macrophages[73]. In humans, ApoE is highly polymorphic, and a relatively frequent isoform (ApoE-4 isoform[74]) causes defective uptake of liver-derived lipoproteins and plasma cholesterol increase; is associated with inflammation; increases phagocytosis and foam cell formation; alters efferocytotic activity; increases antigen presentation potential[75]; and favors fibrous cap thinning due to activation of metalloprotease expression[76]. In addition, ApoE deficiency in mice translates into an increased distribution of GM-CSF receptors by hematopoietic stem cells in the bone marrow, leading to increased and inflammatory myelopoiesis[77,78].
Angptl3 is another protein component of liver-derived lipoproteins which recently appears to be involved in inflammatory mechanisms. Angptl3 physiologically inhibits LPL and EL and, when
Reduced medullary hematopoietic homing was found in Angptl3-null mice[80], a finding not confirmed by Angptl3 gene editing on hematopoietic stem cells in hypercholesteremic mice (on a hypercholesterolemia background due to LDL receptor deficiency[81]). By contrast, hematopoietic stem cells transplanted in Angptl3-null recipients exhibited impaired repopulation[80,82]. Hence, although a tropism for the development of EPCs has peculiarly not been elucidated, it is plausible that Angptl3 acts as regulator of the hematopoietic stemness, dependent on the quantity of liver-derived lipoproteins.
LOWERING LIVER-DERIVED LIPOPROTEINS TO REDUCE INFLAMMATION: WHICH EVIDENCE SO FAR?
Statins (inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A, a rate-limiting step of cholesterol synthesis) and fibrates [agonists of peroxisome proliferator-activated receptor alpha (PPAR-α) that reduce
Statins consistently reduce by up to 15% the basal TG levels per each -50% in LDL-C content in every population and degree of residual CVD risk they have been tested (merely because of a mass effect in reducing the quantity of LDL which carries this TG quantity per particle[83]). Consequently, the efficacy of statins in metabolic liver disease is indisputable. In addition, statins exert the same beneficial role in secondary prevention of CVD, being an optimal start of treatment for patients with metabolic liver disease with still clinical manifestation of atherosclerosis. In addition, statins are safe for patients with different degrees of NAFLD and MAFLD, starting from those with mild baseline elevation in transaminases (< 3× upper limit of normal (ULN)) up to those with compensated cirrhosis[84-89]. Some evidence is available also regarding the potential benefit of statins in reducing the degree of liver fibrosis[90], although more data are needed.
Besides statins, fibrates effectively lower TG across the range of TG levels but only modestly reduce ApoB levels. In addition the cholesterol reduction of fibrates differs between moderate hypertriglyceridemia (TG 150-500 mg/dL, where LDL-C is within normal ranges according to guidelines) and severe hypertriglyceridemia (TG 500-880 mg/dL, where LDL-C is increased, albeit from a low baseline level[24,91,92]). Bezafibrate and fenofibrate demonstrated beneficial effects on both lipid metabolism and liver function in patients with advanced metabolic liver disease and NASH. By contrast to statins, however, the atheroprotective effect of fibrates has been more questioned. More probable beneficial effects in liver histology have been indicated for fibrates. In fact, fenofibrate treatment in patients with advanced MAFLD and NASH decreased transaminases together with hepatocellular ballooning evaluated by biopsy; by contrast, short-term treatment with bezafibrate appeared to reduce microvesicular steatosis[93]. Conversely, no beneficial effects in reducing local tissue inflammation and fibrosis were reported following treatment with fibrates. In addition, the atheroprotective effect of fibrates has been generally questioned. Indeed, while experimental data show mice models of atherosclerosis treated with fibrates benefit from reduced monocyte/macrophage infiltration in the atheroma[94], less is clear regarding data in humans[95,96]. The ACCORD study, combining the use of fenofibrate with simvastatin, failed in providing evidence of an
Apart from these classical strategies and the pemafibrate experience, further options to reduce the burden of liver-derived lipoproteins are currently in the pipeline, harnessing forefront biotechnological techniques[9].
Inhibiting liver-derived lipoprotein production
Drugs such as mipomersen [an antisense oligonucleotide (ASO) inhibitor of ApoB translation] and lomitapide [an inhibitor of microsomal triglyceride transport protein (MTTP) activity] block either ApoB synthesis or the addition of lipid during chylomicron and VLDL assembly in the intestine and liver, respectively. However, the initial clinical use of these compounds found that they both promote hepatic TG accumulation and possible development of NAFLD. Despite representing a valuable alternative in rare severe statin resistant hypercholesterolemia, it is questionable whether their long-term use could be considered in FCHL or the presence of metabolic complications.
Hence, novel or combination therapies that inhibit the assembly of apoB lipoproteins and protect against excess intracellular lipid by promoting FA oxidation or decreasing TG synthesis are needed.
Reducing TG availability for VLDL assembly
High-dose omega-3 FA (3-4 g/day, usually the combination of DHA and EPA) reduces TG and apoB secretion by ~25%-30% and promotes to a very variable extent the peripheral catabolism[103,104]. However, some studies showed an increase in conversion of VLDL to their remnants and LDL; hence, omega-3 FA may have limited impact on remnant populations or the total number of atherogenic lipoproteins.
By contrast, a new formulation of high-dose EPA (icosapent ethyl) induced potent reduction of CVD risk in high-risk patients still on aggressive ongoing statin treatment[105]. Of note, in the same trial, icosapent ethyl provided up to -37% reduction at the end of a 5-year follow-up in high-sensitivity C-reactive protein (CRP), a liver-derived marker of systemic inflammation. In secondary analysis of the same trial, icosapent ethyl was effective in reducing the risk of CVD independently from the presence of diabetes and in the presence of atherogenic dyslipidemia, supporting its potential consideration in NAFLD/MAFLD. Besides, it is questioned whether the actual effect of this EPA formulation was more likely induced by the reduction in liver production of ApoB lipoproteins rather than the peripheral catabolism of their remnants and LDL. Kinetic studies regarding the catabolism of these fractions are required.
Enhancing the peripheral clearance of liver-derived lipoproteins
This option is actually not addressed by fibrates, which, despite promoting the expression and activity of LPL, failed to provide benefit in reducing inflammation and CVD risk[97,98].
The inhibition of ApoCIII and Angplt3, two important down-regulators of LPL activity on the surface of liver-derived lipoproteins, have more recently been addressed by pharmacological research as promising alternatives. An ASO targeting APOC3 gene markedly reduced plasma TG levels in severe hypertriglyceridemia, an effect equally evident in the absence of LPL activity[106,107].
More robust reductions on TG, cholesterol, and ApoB plasma levels have been reached via the inhibition of Angptl3, by both monoclonal antibodies[108] and ASO therapy reducing the synthesis of the protein in patients with metabolic complications and liver disease[109]. Of note, regarding the second option, the robust effect on TG levels in patients with hepatic steatosis was not associated with rebound accumulation of TG in the liver, in contrast to other biotechnological drugs directed to alternative targets (e.g., mipomersen and lomitapide). Long-term studies addressing the anti-atherosclerotic potential of Angplt3 inhibition will be seminal to provide evidence for the hypothesis of this therapeutic strategy in high-risk patients with metabolic complications, including NAFLD/MAFLD.
Further pharmacological options and drugs in the pipeline
Glucagon-like peptide-1 (GLP-1), an incretin hormone produced by intestinal L cells and the brain, is physiologically released during the postprandial phase to stimulate glucose-dependent insulin secretion by the pancreas. The agonism of its receptor (GLP-1R) has been considered in diabetology, with successful data regarding atheroprotection[110]. Additionally, given the less efficient activity of insulin in regulating the excessive secretion of intestinal and liver-derived lipoproteins in diabetes[111], there is a rationale in conceiving such strategy to reduce atherogenic dyslipidemia. Pre-clinical data in rodents demonstrate that GLP1-R agonism reduces VLDL production and hepatic steatosis in addition to an improvement of glycemic control[112]. This paved the road during the last years to consider this option for the treatment of atherogenic dyslipidemia in NAFLD/MAFLD as well. Recent metanalysis of different trials with up to 26 weeks of follow-up (testing multiple GLP1-R agonists, namely liraglutide, exenatide, dulaglutide, and semaglutide) concluded that increasing the signaling of GLP1 resulted in reductions in the absolute percentage of liver fat content (magnetic resonance-based techniques), lowered serum liver enzymes, and improved histology of NASH, without worsening the degree of fibrosis[113]. Further data are needed regarding the effect on lipid profile, as this would further strengthen the relevance of targeting the excessive production of liver-derived lipoproteins to control metabolic liver disease.
Metformin, by increasing AMP-activated protein kinase (AMPK), has been shown to inhibit the differentiation of monocyte to pro-inflammatory macrophages and blunt cytokines secretion by reducing signal transducer and activator of transcription 3 (STAT3) activity. As AMPK is essential for brown adipose tissue (BAT) development and homeostasis, metformin was demonstrated to increase liver-derived lipoproteins uptake and lipolysis by BAT[114].
LXR agonism, by virtue of its presumable effect on liver-derived lipoproteins uptake by the periphery, has been tested. Although LXR agonists exhibited anti-inflammatory effects in the pre-clinical setting, inducing monocytes egression from atherosclerotic plaque[115] and subsequent reduction of plaque size, this option resulted in increased hepatic steatosis, both in healthy subjects and in statin-treated hypercholesterolemic patients[116], questioning the translation in vivo.
Obeticholic acid, an FXR agonist promoting the re-cycle of the cholesterol pool in the entero-hepatic circulation, provided encouraging data in a phase III trial[117] regarding the regulation of liver fibrosis with up to 38% of patients in the 25 mg arm displaying improvement of biopsy-proven fibrosis, even in the presence of metabolic complication. Despite the withdrawal of the compound being announced by the developing company[118], from a lipidological point of view, obetichollic acid induced questionable effects on TG (not consistently reduced as a function of FXR agonism) and LDL cholesterol content (which increased as compared to placebo).
CONCLUSION
The number of subjects who are at risk of liver disease is expected to grow over the coming years and will determine a serious escalation in the incidences of fatal and non-fatal atherosclerotic cardiovascular diseases[119]. This rate will be increased by the elevated pressure of additional comorbidities, with type 2 diabetes and metabolic syndrome in primis[120] constantly increasing in affluent societies with unhealthy lifestyles. Together, these well-described projections call for the worldwide guidelines to determine algorithms for coordinated methods of interventions. At the same time, all the pharmacological trials targeting liver-derived lipoproteins provide a wealth of evidence supporting that the lipid content of the liver is causal for the evolution of CVD risk. In addition, data from these trials are paralleled to those from forefront trials, posing inflammation per se as the real culprit. Under this perspective, for example, PCSK9 monoclonal antibody reduced LDL cholesterol (FOURIER trial[121]) and advanced EPA formulations (REDUCE-IT trial[105]) reduced VLDL quantity and cholesterol, together with convincing atherosclerotic CVD risk in high-risk patients and subjects with advanced metabolic complications including liver disease. A similar degree of benefit was not achieved in the same comorbid patients by neither anti-Interleukin Beta (IL-1b) monoclonal antibody (canakinumab[122]) nor colchicine[123], despite potently reducing a large set of inflammatory markers[124]. Lessons from the critical comparison of these trials[125] will improve the sensitization and understanding of the pathophysiological mechanisms involving dyslipidemias and elevated ApoB liver-derived lipoprotein levels. The pre-clinical evidence regarding their causal effects in the evolution of atherosclerosis and related inflammation will certainly help to foster the sensitization of physicians and pharmacological research and companies in the development of new and more effective therapeutic weapons.
DECLARATIONS
Authors’ contributionsDesigned, wrote and edited the manuscript: Baragetti A
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe work of A. Baragetti is supported by Ministry of Health - Ricerca Corrente - IRCCS MultiMedica; “Cibo, Microbiota, Salute” by “Vini di Batasiolo S.p.A” AL_RIC19ABARA_01; a research award (2021) from “the Peanut Institute”.
Conflict of interestThe author declared that there are not conflicts of interest.
Ethics approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med 2010;363:1341-50.
2. Fracanzani AL, Tiraboschi S, Pisano G, et al. Progression of carotid vascular damage and cardiovascular events in non-alcoholic fatty liver disease patients compared to the general population during 10 years of follow-up. Atherosclerosis 2016;246:208-13.
4. Eslam M, Sanyal AJ, George J. International Consensus Panel. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020;158:1999-2014.e1.
5. Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med 2014;371:1131-41.
6. Adiels M, Olofsson SO, Taskinen MR, Borén J. Diabetic dyslipidaemia. Curr Opin Lipidol 2006;17:238-46.
7. Adiels M, Taskinen MR, Borén J. Fatty liver, insulin resistance, and dyslipidemia. Curr Diab Rep 2008;8:60-4.
8. Björnson E, Adiels M, Taskinen MR, Borén J. Kinetics of plasma triglycerides in abdominal obesity. Curr Opin Lipidol 2017;28:11-8.
9. Bellosta S, Rossi C, Alieva AS, Catapano AL, Corsini A, Baragetti A. Cholesterol lowering biotechnological strategies: from monoclonal antibodies to antisense therapies. A pre-clinical perspective review. Cardiovasc Drugs Ther 2022; doi: 10.1007/s10557-021-07293-w.
10. Baragetti A, Grejtakova D, Casula M, et al. Proprotein Convertase Subtilisin-Kexin type-9 (PCSK9) and triglyceride-rich lipoprotein metabolism: facts and gaps. Pharmacol Res 2018;130:1-11.
11. Nordestgaard BG. A Test in context: lipid profile, fasting versus nonfasting. J Am Coll Cardiol 2017;70:1637-46.
12. Masuda D, Yamashita S. Postprandial hyperlipidemia and remnant lipoproteins. J Atheroscler Thromb 2017;24:95-109.
13. Berry SE, Valdes AM, Drew DA, et al. Human postprandial responses to food and potential for precision nutrition. Nat Med 2020;26:964-73.
14. Sharrett AR, Heiss G, Chambless LE, et al. Metabolic and lifestyle determinants of postprandial lipemia differ from those of fasting triglycerides: the Atherosclerosis Risk In Communities (ARIC) study. Arterioscler Thromb Vasc Biol 2001;21:275-81.
15. Dubois C, Beaumier G, Juhel C, et al. Effects of graded amounts (0-50 g) of dietary fat on postprandial lipemia and lipoproteins in normolipidemic adults. Am J Clin Nutr 1998;67:31-8.
16. Pirillo A, Norata GD, Catapano AL. Postprandial lipemia as a cardiometabolic risk factor. Curr Med Res Opin 2014;30:1489-503.
17. FoodData Central [Internet]. Available from: https://fdc.nal.usda.gov/ [Last accessed on 27 Jun 2022].
18. BDA | Food Composition Database for Epidemiological Studies in Italy [Internet]. Available from: http://www.bda-ieo.it/wordpress/en/ [Last accessed on 27 Jun 2022].
19. Sun L, Cai J, Gonzalez FJ. The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat Rev Gastroenterol Hepatol 2021;18:335-347.
20. Hegele RA, Ginsberg HN, Chapman MJ, et al. The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. lancet Diabetes Endocrinol 2014;2:655-666.
21. Johansen CT, Hegele RA. Genetic bases of hypertriglyceridemic phenotypes. Curr Opin Lipidol 2011;22:247-253.
22. Hansen SEJ, Madsen CM, Varbo A, Nordestgaard BG. Low-grade inflammation in the association between mild-to-moderate hypertriglyceridemia and risk of acute pancreatitis: a study of more than 115000 individuals from the general population. Clin Chem 2019;65:321-332.
23. Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J 2020;41:111-188.
24. Ginsberg HN, Packard CJ, Chapman MJ, et al. Triglyceride-rich lipoproteins and their remnants: metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur Heart J 2021;42:4791-4806.
25. Tada H, Nohara A, Inazu A, Mabuchi H, Kawashiri M aki. Remnant lipoproteins and atherosclerotic cardiovascular disease. Clin Chim Acta 2019;490:1-5.
26. Salinas CAA, Chapman MJ. Remnant lipoproteins: are they equal to or more atherogenic than LDL? Curr Opin Lipidol 2020;31:132-139.
27. Nakajima K, Nakano T, Tokita Y, et al. Postprandial lipoprotein metabolism: VLDL vs chylomicrons. Clin Chim Acta 2011;412:1306-1318.
28. Sniderman AD. Differential response of cholesterol and particle measures of atherogenic lipoproteins to LDL-lowering therapy: implications for clinical practice. J Clin Lipidol 2008;2:36-42.
29. Ference BA, Kastelein JJP, Ginsberg HN, et al. Association of genetic variants related to CETP inhibitors and statins with lipoprotein levels and cardiovascular risk. JAMA 2017;318:947-956.
30. Bini S, D’Erasmo L, Di Costanzo A, Minicocci I, Pecce V, Arca M. The interplay between angiopoietin-Like proteins and adipose tissue: another piece of the relationship between adiposopathy and cardiometabolic diseases? Int J Mol Sci 2021;22:1-16.
31. Minicocci I, Santini S, Cantisani V, et al. Clinical characteristics and plasma lipids in subjects with familial combined hypolipidemia: a pooled analysis. J Lipid Res 2013;54:3481-3490.
32. Ekstedt M, Hagström H, Nasr P, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015;61:1547-1554.
33. Angulo P, Kleiner DE, Dam-Larsen S, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389-397.
34. Tsutsumi T, Eslam M, Kawaguchi T, et al. MAFLD better predicts the progression of atherosclerotic cardiovascular risk than NAFLD: generalized estimating equation approach. Hepatol Res 2021;51:1115-1128.
36. Moreton JR. Physical state of lipids and foreign substances producing atherosclerosis. Science 1948;107:371-373.
37. Schwartz EA, Reaven PD. Lipolysis of triglyceride-rich lipoproteins, vascular inflammation, and atherosclerosis. Biochim Biophys Acta 2012;1821:858-866.
38. Higgins LJ, Rutledge JC. Inflammation associated with the postprandial lipolysis of triglyceride-rich lipoproteins by lipoprotein lipase. Curr Atheroscler Rep 2009;11:199-205.
39. Ting HJ, Stice JP, Schaff UY, et al. Triglyceride-rich lipoproteins prime aortic endothelium for an enhanced inflammatory response to tumor necrosis factor-α. Circ Res 2007;100:381-390.
40. Zewinger S, Reiser J, Jankowski V, et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol 2020;21:30-41.
41. Doi H, Kugiyama K, Oka H, et al. Remnant lipoproteins induce proatherothrombogenic molecules in endothelial cells through a redox-sensitive mechanism. Circulation 2000;102:670-676.
42. De Sousa JC, Soria C, Ayrault-Jarrier M, et al. Association between coagulation factors VII and X with triglyceride rich lipoproteins. J Clin Pathol 1988;41:940-944.
43. Mattavelli E, Catapano AL, Baragetti A. Molecular immune-inflammatory connections between dietary fats and atherosclerotic cardiovascular disease: which translation into clinics? Nutrients 2021:13.
44. Ajuwon K, Spurlock M. Palmitate activates the NF-kappaB transcription factor and induces IL-6 and TNFalpha expression in 3T3-L1 adipocytes. J Nutr 2005;135:1841-1846.
45. Lee J, Zhao L, Youn H, et al. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem 2004;279:16971-16979.
46. Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci 2021;78:1233-1261.
47. Lee JY, Plakidas A, Lee WH, et al. Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res 2003;44:479-486.
48. Veglia F, Tyurin VA, Blasi M, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019;569:73-78.
49. Zheng S, Ma M, Li Z, et al. Posttreatment of Maresin1 Inhibits NLRP3 inflammasome activation via promotion of NLRP3 ubiquitination. FASEB J 2020;34:11944-11956.
50. Viola JR, Lemnitzer P, Jansen Y, et al. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ Res 2016;119:1030-1038.
51. Adam AC, Lie KK, Moren M, Skjærven KH. High dietary arachidonic acid levels induce changes in complex lipids and immune-related eicosanoids and increase levels of oxidised metabolites in zebrafish (Danio rerio). Br J Nutr 2017;117:1075-1085.
52. Holladay CS, Wright RM, Spangelo BL. Arachidonic acid stimulates interleukin-6 release from rat peritoneal macrophages in vitro: evidence for a prostacyclin-dependent mechanism. Prostaglandins Leukot Essent Fatty Acids 1993;49:915-922.
53. Shen Z, Ma Y, Ji Z, et al. Arachidonic acid induces macrophage cell cycle arrest through the JNK signaling pathway. Lipids Health Dis 2018;17:26.
54. Wellenstein MD, de Visser KE. Fatty acids corrupt neutrophils in cancer. Cancer Cell 2019;35:827-829.
55. Alsina-Sanchis E, Mülfarth R, Moll I, Mogler C, Rodriguez-Vita J, Fischer A. Intraperitoneal oil application causes local inflammation with depletion of resident peritoneal macrophages. Mol Cancer Res 2021;19:288-300.
56. Christ A, Lauterbach M, Latz E. Western diet and the immune system: an inflammatory connection. Immunity 2019;51:794-811.
57. Astrup A, Magkos F, Bier DM, et al. Saturated fats and health: a reassessment and proposal for food-based recommendations: JACC state-of-the-art review. J Am Coll ;76:844-857.
58. Sheedy FJ, Grebe A, Rayner KJ, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol 2013;14:812-820.
59. Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol 2015;15:104-116.
60. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010;464:1357-1361.
61. Bekkering S, Quintin J, Joosten LAB, Van Der Meer JWM, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol 2014;34:1731-1738.
62. Christ A, Günther P, Lauterbach MAR, et al. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell 2018;172:162-175.e14.
63. Toutouzas K, Skoumas J, Koutagiar I, et al. Vascular inflammation and metabolic activity in hematopoietic organs and liver in familial combined hyperlipidemia and heterozygous familial hypercholesterolemia. J Clin Lipidol 2018;12:33-43.
64. Bernelot Moens SJ, Verweij SL, Schnitzler JG, et al. Remnant cholesterol elicits arterial wall inflammation and a multilevel cellular immune response in humans. Arterioscler Thromb Vasc Biol 2017;37:969-975.
65. Raposeiras-Roubin S, Rosselló X, Oliva B, et al. Triglycerides and residual atherosclerotic risk. J Am Coll Cardiol 2021;77:3031-3041.
66. Chait A, Ginsberg HN, Vaisar T, Heinecke JW, Goldberg IJ, Bornfeldt KE. Remnants of the triglyceride-rich lipoproteins, diabetes, and cardiovascular disease. Diabetes 2020;69:508-516.
67. Borén J, John Chapman M, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2020;41:2313-2330.
68. Mahely R, Innerarity T, Rall S, Weisgraber K. Lipoproteins of special significance in atherosclerosis: insights provided by studies of type III hyperlipoproteinemia. Ann N Y Acad Sci 1985;454:209-221.
69. Norata GD, Grigore L, Raselli S, et al. Post-prandial endothelial dysfunction in hypertriglyceridemic subjects: molecular mechanisms and gene expression studies. Atherosclerosis 2007;193:321-327.
70. Schnitzler JG, Moens SJB, Tiessens F, et al. Nile red quantifier: a novel and quantitative tool to study lipid accumulation in patient-derived circulating monocytes using confocal microscopy. J Lipid Res 2017;58:2210-2219.
71. Klop B, Proctor SD, Mamo JC, Botham KM, Castro Cabezas M. Understanding postprandial inflammation and its relationship to lifestyle behaviour and metabolic diseases. Int J Vasc Med 2012;2012:1-11.
72. Aboumsallem JP, Muthuramu I, Mishra M, De Geest B. Cholesterol-lowering gene therapy prevents heart failure with preserved ejection fraction in obese type 2 diabetic mice. Int J Mol Sci 2019;20:2222.
73. Pourcet B, Staels B. Alternative macrophages in atherosclerosis: not always protective! J Clin Invest 2018;128:910-912.
74. Rasmussen KL, Tybjærg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Plasma apolipoprotein E levels and risk of dementia: a Mendelian randomization study of 106,562 individuals. Alzheimers Dement 2018;14:71-80.
75. Bonacina F, Coe D, Wang G, et al. Myeloid apolipoprotein E controls dendritic cell antigen presentation and T cell activation. Nat Commun 2018;9:3083.
76. Liberale L, Dallegri F, Montecucco F, Carbone F. Pathophysiological relevance of macrophage subsets in atherogenesis. Thromb Haemost 2017;117:7-18.
77. Westerterp M, Murphy AJ, Wang M, et al. Deficiency of ATP-binding cassette transporters A1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ Res 2013;112:1456-65.
78. Baragetti A, Bonacina F, Catapano AL, Norata GD. Effect of lipids and lipoproteins on hematopoietic cell metabolism and commitment in atherosclerosis. Immunometabolism 2021:3.
79. Kaplan R, Zhang T, Hernandez M, et al. Regulation of the angiopoietin-like protein 3 gene by LXR. J Lipid Res 2003;44:136-143.
80. Zheng J, Huynh HD, Umikawa M, Silvany R, Zhang CC. Angiopoietin-like protein 3 supports the activity of hematopoietic stem cells in the bone marrow niche. Blood 2011;117:470-479.
81. Chadwick AC, Evitt NH, Lv W, Musunuru K. Reduced blood lipid levels with in vivo CRISPR-Cas9 base editing of ANGPTL3. Circulation 2018;137:975-977.
82. Zhang CC, Kaba M, Ge G, et al. Angiopoietin-like proteins stimulate ex vivo expansion of hematopoietic stem cells. Nat Med 2006;12:240-245.
83. Collaboration CTT (CTT). Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170 000 participants in 26 randomised trials. Lancet 2010;376:1670-1681.
84. Athyros VG, Alexandrides TK, Bilianou H, et al. The use of statins alone, or in combination with pioglitazone and other drugs, for the treatment of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis and related cardiovascular risk. An Expert Panel Statement. Metabolism 2017;71:17-32.
85. Chalasani N, Aljadhey H, Kesterson J, Murray MD, Hall SD. Patients with elevated liver enzymes are not at higher risk for statin hepatotoxicity. Gastroenterology 2004;126:1287-1292.
86. Cohen DE, Anania FA, Chalasani N. An assessment of statin safety by hepatologists. Am J Cardiol 2006;97:77C-81C.
87. Kaplan DE, Serper MA, Mehta R, et al. Effects of hypercholesterolemia and statin exposure on survival in a large national cohort of patients with cirrhosis. Gastroenterology 2019;156:1693-1706.e12.
88. Kim RG, Loomba R, Prokop LJ, Singh S. Statin use and risk of cirrhosis and related complications in patients with chronic liver diseases: a systematic review and meta-analysis. Clin Gastroenterol Hepatol 2017;15:1521-1530.
89. Thomson MJ, Serper M, Khungar V, et al. Prevalence and factors associated with statin use among patients with nonalcoholic fatty liver disease in the TARGET-NASH study. Clin Gastroenterol Hepatol 2022;20:458-460.
90. Torres-Peña JD, Martín-Piedra L, Fuentes-Jiménez F. Statins in non-alcoholic steatohepatitis. Front Cardiovasc Med 2021;8:777131.
91. Shepherd J, Caslake MJ, Lorimer AR, Vallance BD, Packard CJ. Fenofibrate reduces low density lipoprotein catabolism in hypertriglyceridemic subjects. Arteriosclerosis 1985;5:162-168.
92. Ginsberg HN. Changes in lipoprotein kinetics during therapy with fenofibrate and other fibric acid derivatives. Am J Med 1987;83:66-70.
93. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol 2015;62:720-733.
94. Kooistra T, Verschuren L, De Vries-Van Der Weij J, et al. Fenofibrate reduces atherogenesis in ApoE*3Leiden mice: evidence for multiple antiatherogenic effects besides lowering plasma cholesterol. Arterioscler Thromb Vasc Biol 2006;26:2322-30.
95. Okopień B, Krysiak R, Herman ZS. Effects of short-term fenofibrate treatment on circulating markers of inflammation and hemostasis in patients with impaired glucose tolerance. J Clin Endocrinol Metab 2006;91:1770-1778.
96. Krysiak R, Gdula-Dymek A, Okopieñ B. Monocyte-suppressing effect of high-dose metformin in fenofibrate-treated patients with impaired glucose tolerance. Pharmacol Rep 2013;65:1311-1316.
97. Ginsberg H, Elam M, Lovato L, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010;362:1563-1574.
98. Scott R, O’Brien R, Fulcher G, et al. Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care 2009;32:493-498.
99. Deprince A, Haas JT, Staels B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol Metab 2020;42:101092.
100. Seko Y, Yamaguchi K, Umemura A, et al. Effect of pemafibrate on fatty acid levels and liver enzymes in non-alcoholic fatty liver disease patients with dyslipidemia: a single-arm, pilot study. Hepatol Res 2020;50:1328-1336.
101. Hatanaka T, Kakizaki S, Saito N, et al. Impact of pemafibrate in patients with hypertriglyceridemia and metabolic dysfunction-associated fatty liver disease pathologically diagnosed with non-alcoholic steatohepatitis: a retrospective, single-arm study. Intern Med 2021;60:2167-2174.
102. Phase 3 CV outcomes study with pemafibrate stopped early - PACE-CME [Internet]. Available from: https://pace-cme.org/2022/04/11/phase-3-cv-outcomes-study-with-pemafibrate-stopped-early/ [Last accessed on 27 Jun 2022].
103. Shearer GC, Savinova O V, Harris WS. Fish oil -- how does it reduce plasma triglycerides? Biochim Biophys Acta 2012;1821:843-851.
104. Oscarsson J, Hurt-Camejo E. Omega-3 fatty acids eicosapentaenoic acid and docosahexaenoic acid and their mechanisms of action on apolipoprotein B-containing lipoproteins in humans: a review. Lipids Health Dis 2017;16:149.
105. Bhatt DL, Steg PG, Miller M, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med 2019;380:11-22.
106. Gaudet D, Brisson D, Tremblay K, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med 2014;371:2200-06.
107. Witztum JL, Gaudet D, Freedman SD, et al. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N Engl J Med 2019;381:531-542.
108. Ahmad Z, Banerjee P, Hamon S, et al. Inhibition of angiopoietin-like protein 3 with a monoclonal antibody reduces triglycerides in hypertriglyceridemia. Circulation 2019;140:470-486.
109. Gaudet D, Karwatowska-Prokopczuk E, Baum SJ, et al. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridaemia. Eur Heart J 2020;41:3936-3945.
110. Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. Drug Ther Bull 2016;54:101.
111. Nogueira JP, Maraninchi M, Béliard S, et al. Absence of acute inhibitory effect of insulin on chylomicron production in type 2 diabetes. Arterioscler Thromb Vasc Biol 2012;32:1039-1044.
112. Parlevliet ET, Wang Y, Geerling JJ, et al. GLP-1 receptor activation inhibits VLDL production and reverses hepatic steatosis by decreasing hepatic lipogenesis in high-fat-fed APOE*3-Leiden mice. PLoS One 2012;7:e49152.
113. Mantovani A, Petracca G, Beatrice G, Csermely A, Lonardo A, Targher G. Glucagon-Like peptide-1 receptor agonists for treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: an updated meta-analysis of randomized controlled trials. Metabolites 2021;11:1-13.
114. Geerling JJ, Boon MR, Van Der Zon GC, et al. Metformin lowers plasma triglycerides by promoting VLDL-triglyceride clearance by brown adipose tissue in mice. Diabetes 2014;63:880-891.
115. Rigamonti E, Chinetti-Gbaguidi G, Staels B. Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler Thromb Vasc Biol 2008;28:1050-1059.
116. Kirchgessner TG, Sleph P, Ostrowski J, et al. Beneficial and adverse effects of an LXR agonist on human lipid and lipoprotein metabolism and circulating neutrophils. Cell Metab 2016;24:223-233.
117. Younossi ZM, Ratziu V, Loomba R, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019;394:2184-2196.
118. Intercept announces withdrawal of EMA marketing authorization application for obeticholic acid for advanced liver fibrosis due to NASH - intercept pharmaceuticals, Inc. Available from: https://ir.interceptpharma.com/news-releases/news-release-details/intercept-announces-withdrawal-ema-marketing-authorization [Last accessed on 27 Jun 2022].
119. Lazarus JV, Mark HE, Anstee QM, et al. Advancing the global public health agenda for NAFLD: a consensus statement. Nat Rev Gastroenterol Hepatol 2021;19:60-78.
120. Diabetes [Internet]. Available from: https://www.who.int/health-topics/diabetes#tab=tab_1 [Last accessed on 27 Jun 2022].
121. Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med 2017;376:1713-1722.
122. Ridker PM, Everett BM, Thuren T, et al. CANTOS Trial Group. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119-31.
123. Bouabdallaoui N, Blondeau L, Tardif JC. Lessons from COLCOT and LoDoCo2: colchicine for secondary prevention in coronary artery disease. Eur Heart J 2021;42:2800-1.
124. Opstal TSJ, Hoogeveen RM, Fiolet ATL, et al. Colchicine attenuates inflammation beyond the inflammasome in chronic coronary artery disease: a LoDoCo2 proteomic substudy. Circulation 2020;142:1996-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Baragetti A. Liver-derived lipoproteins and inflammation: from pathophysiology to pharmacological targets in metabolic liver disease. Metab Target Organ Damage 2022;2:9. http://dx.doi.org/10.20517/mtod.2022.09
AMA Style
Baragetti A. Liver-derived lipoproteins and inflammation: from pathophysiology to pharmacological targets in metabolic liver disease. Metabolism and Target Organ Damage. 2022; 2(3): 9. http://dx.doi.org/10.20517/mtod.2022.09
Chicago/Turabian Style
Baragetti, Andrea. 2022. "Liver-derived lipoproteins and inflammation: from pathophysiology to pharmacological targets in metabolic liver disease" Metabolism and Target Organ Damage. 2, no.3: 9. http://dx.doi.org/10.20517/mtod.2022.09
ACS Style
Baragetti, A. Liver-derived lipoproteins and inflammation: from pathophysiology to pharmacological targets in metabolic liver disease. Metab Target Organ Damage. 2022, 2, 9. http://dx.doi.org/10.20517/mtod.2022.09
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 31
likes
Like This Article 31
likes







Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.